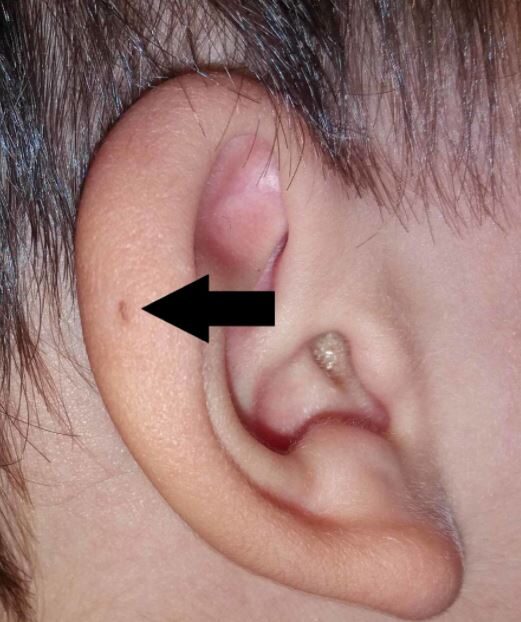
In the first half of the 20th century, PAuS became known as the “natural earring hole” and “fistula auris congenita” with several authors publishing on the topic, including models of inheritance [3,15,16]. The phenomenon and its intergenerational inheritance have allowed for an extensive series of studies and theories on its causes which are largely in line with Virchow’s original claims [17-23].
One of the most important contributions of this period came from Fourman and Fourman in 1955, who researched PAuS inheritance, created family trees, and focused on structural inheritance [24]. They concluded that half of the siblings in the affected family had the structure and therefore postulated that sinus was passed down as a dominant trait, but with “incomplete penetration” as it was passed from generation to generation or individuals [24].
Despite initial interest, however, almost no articles were published after the late 1940s, until a slight resurgence in later years when the structure reappeared under a new name, PAuS. Structural studies have rarely been carried out since then, and their mechanisms of transgenerational inheritance have been largely forgotten in the literature of the 1930s and 1940s.
PAuS . related syndrome
Melnik-Fraser syndrome
This syndrome was first described by Heusinger in 1864. In 1975 Melnick et al. described a number of patients with it and in 1980 Fraser et al. described another case series with syndromic features in special schools for the deaf [25,26]. The disease is considered relatively rare; 250 new cases of Melnick-Fraser were diagnosed in Japan in 2014 [27]. Initially considered a variant of the BOR syndrome, Melnik later expanded the syndrome into a group of nearly identical diseases, depending on the presence or absence of various features [28,29]. Later the term Melnik-Fraser syndrome was introduced to unify the characteristics of the individual syndromes and to fully distinguish them from Fraser syndrome, an isolated urogenital malformation and Fraser-Lynch syndrome, a familial colorectal polyposis with which BOR is often confused separately. Unfortunately, Heusinger’s contributions have been largely forgotten.
Beckwith-Wiedemann syndrome
The condition was first described in 1963 by American pathologist John Beckwith as a combination of exophalos, macroglossia, and gigantism, hence the name Exophalos macroglossia gigantism (EMG) syndrome (Poster: Beckwith, JB: Extreme Cytomegaly of the Adrenal Gland) Renal and Pancreatic Hyperplasia and Leydig – Cellular hyperplasia: another syndrome (Western Society Annual Meeting for Pediatric Research, Los Angeles, California, November 11, 1963). Apart from John Beckwith, German pediatrician Hans-Rudolf Wiedemann also described several patients in 1964 with the same symptoms, but also with symptoms such as adrenal hyperplasia. Over time, the syndrome was referred to as the Bewwith-Wiedemann syndrome and the diagnostic criteria have been expanded to include PAuS, hyperplastic kidney disease, microcephaly, neonatal hypoglycemia, and hepatoblastoma, which develops later in life [31,32]. Mutations have been identified in 11p15 that affect genes such as insulin-like growth factor 2 (IGF-2), cyclin-dependent kinase 1C inhibitor (CDKN1C), H19 and potassium tension, subfamily Q member 1, overlapping transcript 1 (KCNQ1OT) [32] . The mechanism of inheritance has not been determined, because hereditary transgenerational inheritance cannot be precisely determined in some patients with this disease, as it is a sporadic recessive defect. Beckwith-Wiedemann syndrome and its features are critical for differentiation in patients conceived by in vitro fertilization, as its incidence is much higher in this population [33-35]. Beckwith-Wiedemann is considered a rare disease with a frequency of 1 in 13,700 births with a total of about 300 children born with Beckwith-Wiedemann in the United States each year.
Lakhievich-Sibley . syndrome
First published in 1985 by Lachiewicz et al. described, it is one of the rarest syndromes ever described [37]. This syndrome is very similar to Melnick-Fraser, although only PAuS and a hypoplastic kidney with early proteinuria are found in the descendants of British and Irish immigrants who settled in Ohio in the 19th century and later in Nebraska. At the time of the first study of 130 living relatives, 12 members had PauS and a hypoplastic kidney, 10 had only a hypoplastic kidney, and 3 had only PAuS [37]. Although the exact location of the mutation remains unknown, the disease is inherited as an autosomal dominant trait.
Twenty-first century
Currently, PAuS is a common clinical entity with a clear clinical treatment strategy, although it has not been widely studied. Several studies have identified a genetic association between PAuS and the 8q11.1-13.1 locus; however, the results are generally not reproducible [1,5,7,38]. Apart from this single study, no other study has focused on the genetic cause of PauS, nor has it associated it with the above genetic syndromes, and pedigree studies are rarely performed [16,24,39].
The clinical course is well described and treatment strategies, which primarily involve surgical excision, are well established in modern ENT and head and neck surgery [5,7,8,40]. Currently, patients with PauS unassociated with genetic syndromes undergo a safe intervention process with few described complications, while patients with genetic syndromes receive the same treatment modalities and excision of PauS does not affect their overall clinical course [7, 40–42] .
Conclusion
PAuS, as described first, is often a component of several hereditary syndromes of the same name such as Melnik-Fraser (BOR syndrome), Beckwith-Wiedemann and Lachiewicz-Sibley, which also include various permanent kidney disorders and other distant disorders such as cysts, fistulas. . , Facial and nerve defects and others. These relatively rare syndromes emphasize the importance of Virchow’s early assertion that the condition is caused by improper fusion of the pharyngeal arch, a process that occurs in parallel at several stages of kidney development. However, there is no evidence in the scientific literature to suggest that the presence of PAuS always ends in the presence of any of the above syndromes. Although the exact inheritance pattern of PauS according to modern knowledge and its genetic and molecular features have not been fully defined and elucidated, some authors still consider it to be a faulty dominant mutation with reduced penetration.